Background
An avian influenza A(H7N9) virus has emerged in south eastern China in March 2013 [1]. As of 16 April 2013, the Chinese authorities have reported 63 laboratory-confirmed human cases, 14 of whom have died [2]. While epidemiological data suggest no direct human-to-human transmission, there is huge concern that the presence of mutations typical for mammalian-adapted influenza A viruses such as E627K in the polymerase basic protein 2 (PB2) gene might indicate a certain propensity of the virus to further adapt to humans [1,3]. Even in absence of proven human-to-human transmission, the emergence of the avian influenza A(H7N9) virus in humans constitutes a test scenario for pandemic preparedness.
The rapid deployment of diagnostic methodology is among the top priorities in laboratory-based pandemic response. While capacities and responsibilities are in place in many countries, the actual provision of test technology involves major challenges, including the necessity to provide validation data for new test protocols, as well as the need for qualified and safe biological materials suitable as positive controls. In particular, positive controls based on in-vitro transcribed RNA containing only small fragments of the viral genome can be shipped without biosafety concerns. We already started using this option for the wide distribution of diagnostic tests during the severe acute respiratory syndrome (SARS) epidemic in 2003, and made use of it several times thereafter [4-6]. In response to the emergence of HCoV-EMC in 2012 we provided validated protocols along with positive controls through a European Union (EU) research network. This strategy enabled implementation of diagnostic capacity across the EU within only a few weeks [7,8]. In this report we present diagnostic methods for detection of the emerging influenza A(H7N9) virus from clinical specimens.
Methods
Clinical samples and influenza cell culture supernatants
Respiratory swabs, sputum, and endotracheal aspirates were obtained during 2012 and 2013 from hospitalised patients of the University of Bonn Medical Centre and the University of Marburg Medical Centre. Cell culture supernatants from typed influenza viruses were obtained from the German Society for Promotion of Quality Assurance in Medical Laboratories (INSTAND) proficiency testing panels. RNA was extracted from the samples as described earlier by using a viral RNA mini kit (Qiagen) [8].
Template for design of assays
The first three published genome sequences of the 2013 influenza A(H7N9) epidemic from the GISAID EpiFlu database, as listed in Table 1, served as the template for assay design. An influenza A/Mallard/Sweden/91/2002 (H7N9) strain [9], provided by Ron Fouchier, Rotterdam, to author M.M. was used for initial validation experiments.
Table 1. Origin of the haemagglutinin and neuraminidase sequences of emerging influenza A(H7N9) virus used for assay design, April 2013
Real-time reverse-transcription polymerase chain reaction targets
In order to design highly specific real-time reverse-transcription polymerase chain reaction (RT-PCR) targets that would not cross-react with human influenza viruses, we chose the haemagglutinin (HA) and neuraminidase (NA) genes of avian influenza A(H7N9) as targets for amplification.
Because no isolates of the emerging influenza A(H7N9) lineage were available from China, we selected an influenza A/Mallard/Sweden/91/2002 (H7N9) strain whose HA and NA genes were closely related [1,9]. The finding of annealing sites for primers and probes was guided by an alignment of three available sequences from the 2013 emerging influenza A(H7N9) lineage, and the influenza A/Mallard/Sweden/91/2002 (H7N9) sequence. Thermodynamically suitable primers and probes were selected to minimise the number of nucleotide mismatches at their binding sites to the emerging A(H7N9) sequences as well as the A/Mallard/Sweden/91/2002 (H7N9) sequence. The NA gene fragment of A/Mallard/Sweden/91/2002 (A7N9) had to be sequenced for this purpose.
The final test layout included two adjacent regions in the HA gene, termed HA(I) and HA(II), which were respectively targeted by primers and probes of two RT-PCR assays. The two HA regions were included in one control RNA construct derived from the influenza A/Mallard/Sweden/91/2002 (H7N9) strain (Figure 1A). A region was also chosen for amplification of the NA gene, constituting the target of a third RT-PCR assay (NA(I)). A respective control RNA for this NA gene region, derived from the influenza A/Mallard/Sweden/91/2002 (H7N9) strain was also constructed (Figure 1B). In each of the three regions targeted by the RT-PCR assays, mutations in the oligonucleotide binding sites between the emerging influenza A(H7N9) lineage sequences and the influenza A/Mallard/Sweden/91/2002(H7N9) strain sequence were minimal, enabling the use of influenza A/Mallard/Sweden/91/2002 (H7N9)-derived RNAs as positive controls for all RT-PCR assays (Figure 1C and D).
Figure 1. Target sequence regions used for real-time reverse-transcription polymerase chain reaction assays to detect the emerging influenza A(H7N9) virus, March 2013
Real-time reverse-transcription polymerase chain reaction
All three assays had the same conditions but the primer and probe sequences varied (Table 2). A 25-μl reaction was set up containing 5 μl of RNA, 12.5 μl of 2 X reaction buffer provided with the Superscript III one step RT-PCR system with Platinum Taq Polymerase (Invitrogen; containing 0.4 mM of each deoxyribonucleotide triphosphates (dNTP) and 3.2 mM magnesium sulfate), 1 μl of reverse transcriptase/Taq mixture from the kit, 0.4 μl of a 50 mM magnesium sulfate solution (Invitrogen – not provided with the kit), 1 μg of non-acetylated bovine serum albumin (Roche), 400 nM concentrations of each of the primers, as well as 200 nM of the probe. All oligonucleotides were synthesised and provided by Tib-Molbiol, Berlin, where stock solutions from the original synthesis lots are kept. Thermal cycling consisted of 55°C for 15 min, followed by 95°C for 3 min and then 45 cycles of 95°C for 15 s, 58°C for 25 s.
Table 2. Primers and probes for assays used to screen for the emerging influenza A(H7N9) virus, April 2013
In-vitro transcribed RNA controls
Using influenza A/Mallard/Sweden/91/2002 (H7N9) strain RNA as a template, a reverse-transcription PCR fragment encompassing both HA regions respectively targeted by the two HA(I) and (II)assays as well as additional flanking nucleotides was generated using primers IVT_HA-FWD and IVT_HA-REV. Likewise a reverse-transcription PCR fragment comprising the region of the NA gene targeted by the NA(I) assay was amplified with primers IVT_NA-FWD and IVT_NA-REV (Table 2). The HA and NA reverse-transcription PCR fragments are thereafter referred to as ‘peri-amplicon fragments‘. These PCR products were ligated into pCR 4 plasmid vectors and cloned in Escherichia coli by means of a pCR 4-TOPO TA cloning reagent set (Invitrogen). Plasmids were examined for correct orientation of inserts by PCR, purified, and re-amplified with plasmid-specific primers from the reagent set to reduce the plasmid background in subsequent in vitro transcription. Products were transcribed into RNA with the MegaScript T7 in vitro transcription reagent set (Ambion). After DNase I digestion, RNA transcripts were purified with Qiagen RNeasy columns and quantified photometrically. The RNAs derived from the peri-amplicon fragments were used as positive control for the performance of the RT-PCR assays (Figure 1).All transcript dilutions were carried out in nuclease-free water containing 10 μg/mL carrier RNA (Qiagen).
Results
Sensitivity of the real-time reverse-transcription polymerase chain reaction assays
Sensitivity tests employed quantified, in-vitro transcribed RNA derived from the peri-amplicon fragments of the combined HA(I/II) assays, as well as the NA(I) assay. Transcripts were generated and tested in serial tenfold dilution experiments. To obtain a statistically robust assessment of limits of detection (LODs), transcripts were tested in multiple parallel reactions containing RNA copy numbers above and below the pre-determined end point dilution detection limits of each assay. The results in terms of the fractions of positive reactions at each concentration were subjected to probit regression analysis.
Detection probabilities of >95% were achieved at RNA concentrations 7.0 and 7.8 copies per reaction with the HA(I) and NA(I) assays, respectively (Figure 2). Probit analysis is not shown for the HA(II) assay because this assay is not proposed as a first line test; however, sensitivity of this assay was highly comparable to that of HA(I).
Figure 2. Probit regression analyses to determine the sensitivity of the real-time reverse-transcription polymerase chain reaction assays developed to detect the emerging influenza A(H7N9) virus, April 2013 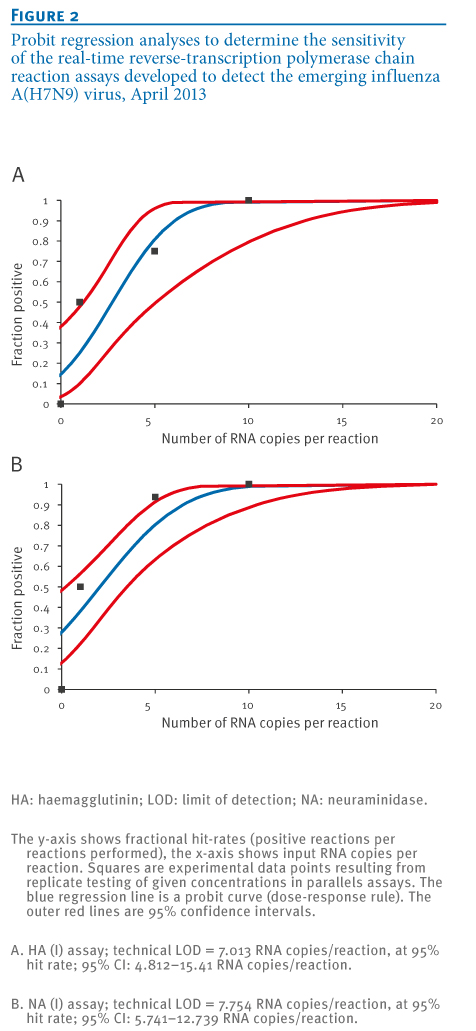
Because the peri-amplicon HA(I) and HA(II) oligonucleotide binding sites each presented with a small number of mismatches to the primers and probes designed for the RT-PCR assays (Figure 1), the sequence of the combined peri-amplicon region of the assays was synthesised in-vitro by PCR fusion of oligonucleotides fragment I and II using primers F and HA7_2_2013rtR to match the A/Anhui/1/2013 sequence (region 1491–1629) 100% (Table 1). The fragment was cloned in E. coli (GenExpress) and transcribed into RNA to be used for parallel testing of the HA assays. For both assays a concentration of five copies of RNA per reaction returned positive in nine of 10 replicates (none were positive with 0 copies per reaction, and all with 50 copies).
Specificity of the assays
To exclude non-specific reactivity of oligonucleotides among each other, all formulations were tested 45 times in parallel with assays containing water and no other nucleic acid except the provided oligonucleotides. In none of these reactions was any positive signal detected. Cross-reactivity with known heterospecific human influenza A viruses as well as other human repiratory viruses was excluded by testing virus positive clinical specimens and high-titre cell culture materials as summarised in Table 3.
Table 3. Known respiratory viruses used for testing the specificity of the assays developed to detect the emerging influenza A(H7N9) virus, April 2013
To obtain a clinically relevant figure of assay specificity, all assays were applied on original clinical samples in which other respiratory viruses had already been detected during routine screening at Bonn and Marburg University Medical Centers (Table 3). These samples were prepared using the Qiagen Viral RNA kit, a formulation widely used to extract RNA in clinical laboratories. Of note, the tested panel included samples containing human influenza A viruses. In total, none of the 121 original clinical samples containing a wide range of respiratory viruses gave any detection signal with either assay, while positive controls were detected. It was concluded that the assay could be applied reliably for clinical samples.
During our validation studies, Word Health Organization (WHO) released RT-PCR protocols targeting other regions of the HA and NA genes (http://www.who.int/influenza/gisrs_laboratory/a_h7n9/en/ on 9 April 2013). Due to the lack of sequence agreement between the A/Anhui/1/2013 (H7N9) and A/Mallard/Sweden/91/2002 (H7N9) we were not able to evaluate the sensitivity of those assays. However, we included them for specificity testing running a panel of clinical samples as listed in Table 3. No false-positive amplifications were encountered while a full validation of these assays would require access to the A/Anhui1/2013 (H7N9) viral RNA or to generate a longer synthetic gene.
Conclusions
Medical laboratories often use conserved target genes such as the matrix gene for the detection of influenza. In cases of suspected human infection with the emerging influenza A (H7N9) strain, however, laboratories need to make sure their diagnostics do not return false positive results due to cross-reactivity with ubiquitous human influenza A viruses. Such cross-reactivity is likely to occur with matrix gene assays, and will thus pose a risk of misleading interpretations of test data. The here-provided protocols provide high specificity for influenza A(H7N9) while detecting minute quantities of virus due to high analytical sensitivity.
In cases of positive detection of influenza A(H7N9), laboratories would want to achieve confirmation by sequence analysis of the amplified fragment. The two primer pairs IVT_HA-FWD, IVT_HA-REV and IVT_NA-FWD, IVT_NA-REV enable sequence confirmation in the HA and NA genes, respectively. It is important to note that the provided in-vitro transcribed RNA controls contain mutations to be discriminated from the emerging influenza A(H7N9) lineage RNA, making it possible to discriminate true virus detections from possible laboratory contaminations. Control material is available from the authors through the European Virus Archive (www.european-virus-archive.com).
Acknowledgements
We gratefully acknowledge the authors, originating and submitting laboratories of the sequences from GISAID’s EpiFlu Database used for assay design (www.gisaid.org).
The development and provision of these assays was done by a European research project on emerging diseases detection and response, EMPERIE, http://www.emperie.eu/emp/, contract No 223498 and the European Union Seventh Framework Programme [FP7/2007-2013] under Grant Agreement n°278433-PREDEMICS (http://predemics.biomedtrain.eu/cms/).
Authors C.D., M.M., M.E. and S.B. has received support from the German Centre for Infection Research (DZIF) that included full funding of the position of author V.M.C. at the Institute of Virology, Bonn. The European Virus Archive platform (www.european-virus-archive.com), through which in-vitro transcribed control RNA for the HA(I), HA(II) and NA(I) assays can be acquired from author C. D is funded by the European Commission under contract number 228292.
Conflict of interest
None declared.
Authors’ contributions
Authors VMC, ME, OL, MM, S Becker and CD designed the study and analysed data. VMC, ME, OL, TB, S Brünink, and MEB did experiments. VMC, ME, OL, MM, S Becker and CD wrote and revised the article.
References